4D imaging via remote refocus
This project is maintained by amsikking in the York lab, and was funded by Calico Life Sciences LLC
Research article
Note that this is a limited PDF or print version; animated and interactive figures are disabled. For the full version of this article, please visit one of the following links: https://amsikking.github.io/remote_refocus https://andrewgyork.github.io/remote_refocus https://calico.github.io/remote_refocus
Remote refocus enables class-leading spatiotemporal resolution in 4D optical microscopy
*Institutional email: amsikking+RR@calicolabs.com †Institutional email: agy+RR@calicolabs.com; Permanent email: andrew.g.york+RR@gmail.com; Group website: andrewgyork.github.io
Abstract
High resolution in space and time is often essential for studying life under a microscope. Fast 2D microscopy has been routine for decades, limited by signal levels or detector speed, but 3D is much slower, typically limited by the focusing method. Here we show that remote refocus [Botcherby 2007] removes focusing as a speed limit while adding minimal drawbacks, and we propose that remote refocus is the right technique for most high-speed high-resolution 3D microscopy applications, especially fluorescence. To our knowledge, there is no commercial implementation of remote refocus, so we provide a modular high-performance design to enable others to build their own. We also present the concept, method and rules of remote refocus to help others design their own. Our design gives camera-limited speed (>4.2x108 voxels/s) and diffraction-limited resolution (<270 nm) over a user-specified volume up to 200x200x60 μm3. We demonstrate the speed and flexibility of remote refocus using a variety of live biological samples with 3D rates ranging from 2.5-50 volumes per second.
Intended audience
Microscopy users, developers, engineers and scientists interested in fast volumetric imaging at high resolution. We tried to write the article in an accessible and straightforward fashion; our aim is to lower the barrier to making a good remote refocus, allowing readers to easily replicate our design, or design and build their own with confidence.
Peer review status
Pre-print published January 11, 2018 (This article is not yet peer-reviewed)
Cite as: doi:10.5281/zenodo.1146083
Introduction
Remote refocus removes the primary speed limit of 3D microscopy, with minimal drawbacks
Revealing the 3D dynamics of living samples is arguably the greatest strength of optical microscopy. To capture these dynamics, we assemble 4D spatiotemporal information from a series of 3D spatial measurements, which are in turn assembled from a series of 2D images [Fischer 2011]. 2D imaging at "video rate" (~30 frames per second) or above has been standard in the field for decades [Inoué 1997], but adding the third dimension with standard focusing methods is comparatively slow [Lukes 2016], and still represents a challenge for those aiming to achieve the fastest volumetric frame rates at the highest resolutions [Bouchard 2015].
Light flux from the sample sets a fundamental limit on temporal resolution; even a detector with infinite speed and zero noise must wait until it detects enough photoelectrons to form an acceptable image [Pawley 2006]. Photoelectron-limited operation is rarely achieved due to hardware limitations, which in 2D imaging is often the camera speed (pixels per second) and in 3D imaging is typically the axial (Z) focusing mechanism.
| Imaging method: | (Differential interference contrast, or transmitted light/fluorescence) |
Many research groups have attacked this focusing speed bottleneck. Notable approaches include imaging multiple focal planes simultaneously via diffractive optics [Blanchard 2000, Prabhat 2004] or light field measurements [Levoy 2006, Broxton 2013], with recent highlights in both approaches [Abrahamsson 2013, Prevedel 2014]. Here we hope to revive a technique of similar vintage, the remote refocus (RR) [Botcherby 2007], and suggest that remote refocusing is the right way to acquire fast, high resolution 3D fluorescent images in most cases.
In our hands and others [Botcherby 2008], RR eliminates the 3D bottleneck, reducing axial focusing time below the dead time between camera frames. Remote refocusing's drawbacks are minimal: lower optical efficiency due to four extra lenses in the emission path, and an extended depth of field at the highest volumetric frame rates. Whilst lower efficiency is never desirable, additional lenses in the emission path are tolerable and typical in many microscopes, such as spinning disk units, external filter wheels, etc. Elongated depth of field is either beneficial or undesirable depending on the application [Botcherby 2008], but only becomes relevant at the highest volumetric rates, when RR piezo settling times exceed the rolling time of our camera (see design section for details).
In contrast, multifocus [Abrahamsson 2013] and light field microscopy [Prevedel 2014] completely eliminate axial focusing time, but also reduce camera-limited imaging speed (i.e. voxels per second). Multifocus microscopy's X, Y, and Z fields of view can't be adjusted to match the sample, which wastes time measuring empty voxels in undersized samples, and requires slow mechanical focusing for oversize samples. Light field microscopy yields even fewer voxels per second than multifocus microscopy, because it measures multiple camera pixels to characterize one Nyquist-limited voxel; other drawbacks include degraded resolution and mandatory post-processing. These techniques are therefore faster than RR when the bottleneck is RR focusing time, but slower when the bottleneck is the pixel rate of the camera. In our experience with well-engineered RR fluorescence microscopes, the bottleneck is always the camera. In addition, multifocal and light field microscopy combine awkwardly with powerful 3D techniques like light-sheet or spinning-disk microscopy, which couple elegantly with remote refocus [Botcherby 2008, Dunsby 2008].
Given the importance of fast 3D microscopy, and the power and flexibility of RR, we're surprised RR isn't more popular. The history of RR shows many clever technical innovations, but few biological applications, and no commercial implementations. We speculate RR suffers from two problems: surprising pitfalls during design and construction, and a lack of advertising. We hope our design empowers builders to confidently make their own RR, and we hope that 3D videos of C. elegans at 20 volumes per second (Figure 1) and flowing yeast at 50 volumes per second (Figure 2) entice microscope users to try RR for their own high-speed imaging challenges. Most of all, we hope to inspire microscope manufacturers to commercialize and disseminate remote refocus as the "gold standard" solution for fast 3D fluorescence microscopy.
The why and how of remote refocus
The motivation for fast focusing is clear, but the advantages (or indeed, the meaning) of ‘remote’ refocusing may not be obvious. Standard microscopes select their image plane (dotted line, Figure 3a) by moving either the objective lens or the sample. This is convenient and simple, but the high inertia of these elements requires a high-power Z-actuator to achieve agile motion, which combines awkwardly with typical crowded multi-objective turrets. In addition, high-resolution imaging requires immersion media like oil or water, which couples rapid vibration of the objective to the coverslip, disturbing the sample and blurring the image at high focusing speeds (Figure 3c). In our experience, any optimization to improve this focusing speed [York 2013, Sup. Fig. 6] is highly dependent on both the sample and the objective lens, and must be re-optimized to maintain high performance whenever either changes.
Refocusing remotely leaves the sample and primary objective stationary (Figure 3b), isolating the sample from mechanical disturbance at high speed (Figure 3d). Because the moving part is now outside the microscope, optimizations for fast focusing become indpendent of the sample and the primary objective, simplifying life for the microscope's user. The 'bolt-on' nature of the module doesn't interfere with the normal operation of the microscope, and even provides a convenient space for a fast external filter wheel without the need for an additional telescope.
A detailed description of RR theory is elegantly presented in the original paper [Botcherby 2007], and will not be re-visited here. We do however provide a straightforward concept, rules and simulation section in the Appendix for those looking to build their own RR, which highlights potential pitfalls. We also give a brief summary of the history of the technique which may help the reader frame its use in the field of microscopy and understand how the RR version presented here fits into the story.
Results
Large volumes with uncompromised resolution
We designed our remote refocus to couple to a typical microscope stand and produce diffraction-limited images using a 60x 1.4 numerical aperture (NA) oil immersion primary objective (Figure 4). Because our design's Z-drive can make small steps (0.1-1 μm) faster than the rolling time of modern sCMOS cameras at full field (<10 ms), the system imaging speed is either camera-limited or photoelectron-limited in this regime. This section describes the optical performance; for a detailed description of the module, see the design and build sections of the Appendix.
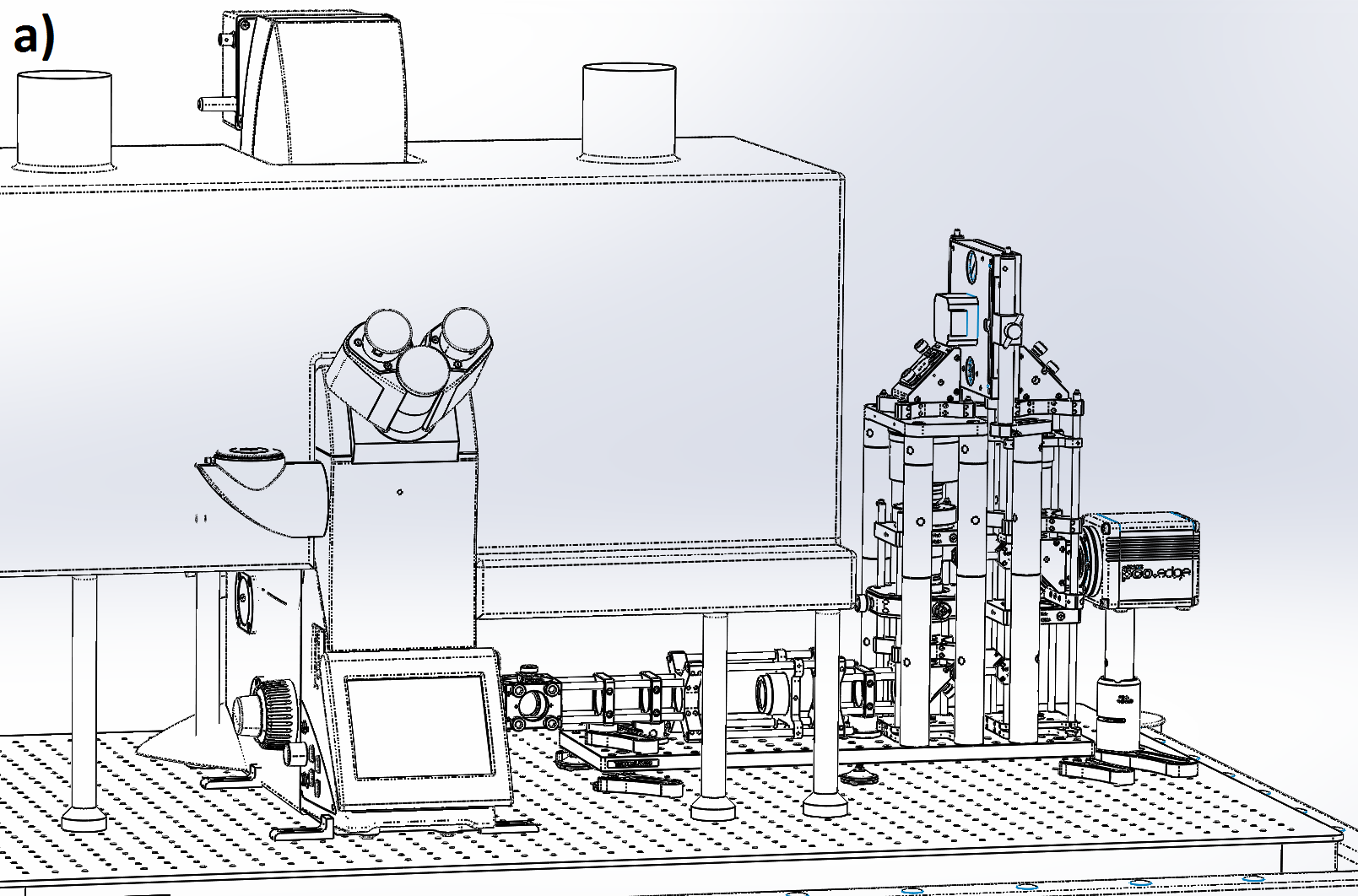


We characterize image quality via Argolight's SIM slide, with variable-spacing fluorescent features (down to 30 nm separation) which allow us to quickly and accurately measure spatial resolution over the full 3D field-of-view. We eschewed traditional labour-intensive volumetric PSF characterisation via beads because of the rigor of the original RR paper [Botcherby 2007], the comprehensive testing of subsequent builders [Anselmi 2011, Qi 2014], and the thorough optical design work implemented here.
Our system yields minimally distorted images across the entire 200x200x60 μm3 volume (Figure 5), using a standard GFP filter cube; note the clearly resolved line pairs, which are separated by 750 nm. We construct a "virtual" 3D test object from a 2D resolution target by moving the microscope's primary objective Z-drive and refocusing via the RR piezo (see the acquisition section for details and raw data).

| Microscope de-focus: | (μm; at 0, the sample is in focus on the microscope eyepieces) | |
| RR de-focus: | (μm; at 0, the RR approximately cancels the primary objective's defocus) |
To quantify resolution, we translated finely spaced resolution lines through the field of view. Consistent with our expections of diffraction-limited performance within the design volume, the RR does not degrade resolution, easily resolving lines separated by 270 nm across the entire volume (Figure 6), and 240 nm near the center (see design section for details). Note the classic Rayleigh limit \(\frac{0.61\lambda}{NA}\) predicts ~230 nm resolution using this filter cube, which even unmodified microscopes rarely achieve in our experience.

| Sample position: | (within the microscope's full field of view) | |
| Microscope de-focus: | (μm; at 0, the sample is in focus on the microscope eyepieces) | |
| RR de-focus: | (μm; at 0, the RR approximately cancels the primary objective's defocus) |
Broadband optical efficiency tests show the RR throughput is >73%, and stability tests show a mechanical drift of ~150 nm/hour (see the design section for details). Finally, Figure 7 explicitly demonstrates volumetric imaging via a 3D rendering of an Argolight structure that consists of an array of rings separated by 5 μm in each axis.
Much faster than standard focusing, especially with non-rigid and delicate samples
Having established excellent image quality, we next measured the speed of our remote refocus (Figure 8) using a series of standard samples: a glass slide with chrome graticule (very rigid), a glass slide with agarose pad and coverslip (semi-rigid), a plastic dish with coverslip bottom (semi-flexible) and a microfluidic chip (very flexible). We compare our RR's focusing speed vs. standard focusing methods: the motorized primary objective of a premium microscope stand, and a high-performance Z-piezo stage insert. For each sample, we set the microscope stand primary objective to an initial 10 μm de-focus, and used each device in turn to focus the sample and then return to its initial position. The allotted focusing times for the RR piezo (15 ms) and the piezo stage insert (50 ms) were determined by their measured 10 μm step-and-settle times (see the acquisition section for details). The focusing time of the motorized objective (~100 ms) is bottlenecked by serial port communication latency. Note that serial port communication is the primary bottleneck for most microscopy systems; we encourage anyone interested in fast acquisition to implement hardware synchronization via an analog-out card.


| Sample choice: |
The graticule is an opaque chrome mask deposited on a small, thick glass slide, supported by a larger solid metal slide. This stiff, well-clamped sample is nearly undisturbed by the immersion oil during fast focusing via the piezo stage insert (50 ms), and it may not be obvious what advantage RR offers in this regime. However, the RR is still >3x faster (15 ms), because RR lets us use a powerful, bulky piezo, tuned to drive a fixed mass. This large tuned piezo is ill-suited for conventional focusing: it doesn't fit typical microscope nosepieces, its power cable antagonizes objective turret rotation, and it would need retuning every time the sample or primary objective changed.
The advantages of RR increase for less rigid samples, which are more typical in our experience. For example, the agarose sample sandwiches C. elegans on a ~1 mm agarose pad between a standard glass slide and a 170 μm coverslip, or the dish sample, a standard manufactured plastic dish with bonded 170 μm coverglass and a sprinkling of aluminium particles for imaging contrast. The flexibility of the agarose prevents the piezo stage insert from reaching sharp focus in the alloted time, and even more so for the dish. Predictably, the remote refocus was unaffected by the flexibility or mass of either sample.
The microfluidic chip is a finely featured polydimethylsiloxane (PDMS) device bonded to a 60x50 mm2 100 μm coverglass, designed for long-term high resolution 3D microscopy of living yeast. The sample is inherently flexible, and moving the microscope objective or stage insert bends it substantially via the immersion oil, requiring more than 400 ms settling time. Of course this is irrelevant to the RR, which gives an impressive 27x speed advantage in this case. This eliminates a common tension between microscope builders and microscope users; it's easy for the builder to criticize a sample for being too flexible, but redesigning a complex sample should not be taken lightly. Our colleagues are delighted that the remote refocus saves them from choosing between slow image acquisition or laborious chip redesign, and we suspect many others would have a similar experience.
Dynamic 4D biology
Having established excellent optical and mechanical performance, we next explore the utility and the limits of remote refocus for imaging living organisms at high spatiotemporal resolution. Samples include C. elegans roaming an agarose pad imaged at up to 20 volumes/s (Figure 1), yeast cells flowing in microfluidic channels imaged at up to 50 volumes/s (Figures 2 and 9), and waterborne protozoa freely moving in a 3D volume imaged at up to 16.7 volumes/s (Figure 9).
| Sample choice: |
RR enables whole-organism DIC microscopy fast enough to 'freeze' the motion of C. elegans pharyngeal pumping (Figure 1 with Imaging method: DIC) or Amoeba proteus internal flow (Figure 9 with Sample choice: Amoeba) in 3D. Of course, fast focusing is necessary but not sufficient for fast 3D microscopy, and we must manage many potential speed limits. For example, adding a fluorescent channel (Figure 1 with Imaging method: TL/FL) requires twice as many camera frames, cutting volumetric rates in half. Since DIC collection optics block ~50% of fluorescent emission, we typically switch to transmitted light (TL) when using fluorescence, rather than lose speed from dim fluorescence or slow mechanical filter changing.
RR combined with fast-switching LED illumination and an appropriate filter set allows high-speed multi-color imaging of rapidly flowing yeast (Figure 9 with Sample choice: Yeast - 3 color). Note that even at 4.6 volumes per second, the flow still moves significantly during each volume. In this case, our speed limit isn't focusing, signal rates, or illumination intensity; it's the camera. Current sCMOS chips measure at most 2x105 lines per second, independent of the number of pixels per line. Cropping our camera to a small number of long lines (Figure 2) enables 50 volumes per second, fast enough to 'freeze' this rapid flow without sacrificing pixels per second, and orienting the flow perpendicular to the camera lines maximizes yeast per second measured by this '3D cytometer'.
Piezo motion potentially elongates the depth of field when the piezo step-and-settle time (~10 ms here) exceeds the camera "dead time" between illuminations (~5 μs per line here). For example, in Figure 2 we cropped the camera vertically to 200 lines, and each 2 ms exposure consists of a 1 ms dead time followed by 1 ms of illumination (500 frames/s). We moved the RR piezo at a constant 1.66 mm/s velocity during each bi-directional volume, extending the effective depth of field by 1.66 μm, ~2 fold the static depth of field estimated via \(\frac{2\lambda n}{\text{NA}^2}\) (~750 nm). We found the resulting Z-resolution acceptable, but there are ways to circumvent the issue, e.g. a larger piezo, a lighter RR objective, a more sophisticated control voltage; perhaps simplest is to use a shorter illumination time.
While searching a Spirostomum sample, we incidentally acquired two interesting videos of "contaminant" species that highlight limits of our design. We imaged the first species (Figure 9 with Sample choice: Contaminant - Epistylis?) at 16.7 volumes/s. While the main body of the creature is almost stationary during one volume, the cilia near the 'mouth' show motion blur during a single 10 ms 2D exposure. This suggests we'd need to image at least 200-300 volumes/s to 'freeze' this rapid motion in 3D, which approaches the resonant frequency of our piezo and the limit of our camera's frame rate for useful fields of view. The second species (Figure 9 with Sample choice: Contaminant - Paramecium?) is even faster, moving its entire body almost this quickly. We suspect simultaneous multiplane methods like multifocus or light-field microscopy would be better suited for imaging such rapid motion; even with volumetric frame rates no faster than RR, at least they could avoid motion blur during one volume by strobing sufficiently bright illumination for a sufficiently short time.
Discussion
Paths towards widespread adoption of remote refocus
We believe that RR is ripe for widespread adoption, cleanly and flexibly addressing a common challenge without introducing other problems. Many microscopes could benefit from decoupling the focusing mechanism from the sample via remote refocus, especially when the sample is sensitive and high spatiotemporal resolution is required. RR's modularity is a great strength; you can treat it as a "black box" attached directly before your camera to eliminate focusing speed limits, without interfering mechanically or optically with other features of your microscope, ranging from objective turrets and emission filters to spinning-disk units or even structured illumination microscopy. To our knowledge, the foundational patent claiming aspects of the RR design [Wilson 2013] is available for licensing, and we encourage microscope vendors to explore this possibility.
For some applications, we would still recommend other approaches. For example, RR enables high volumetric frame rates, but doesn't deliver truly simultaneous "snapshot" volumetric imaging, unlike multifocus or light-field microscopy. Although the hardware cost of our design (~$30k) is a fraction of a high-end fluorescence microscope's price, light-field microscopy's hardware is even cheaper, a simple microlens array (~$1k) and a powerful computer. Of course, our RR's price is dominated by the piezo and objectives, and could be redesigned to minimize cost.
Perhaps the greatest obstacle to building your own RR is getting software to control the hardware. We write our own Python code to control our hardware. This approach is powerful, flexible, and open-source, but our code is poorly documented and very specific to our needs. We encourage anyone interested in learning this approach to contact or visit Andrew York, although we caution that this is only appropriate for people who are proficient in Python programming, or serious about achieving proficiency.
We'd especially like to see a μManager device adapter [Edelstein 2014] for our RR design. Since the controller for our piezo can act as an analog-out card programmed via the serial port, this is potentially straightforward. If other groups share this interest, please contact us; we are not expert μManager hackers, but we may be able to contribute attention and resources to this effort.
Most of all, we want to see the field of microscopy advance, and we're eager to see others adopt remote refocus. If you're considering building your own, we'd enjoy hearing about your plans and your design, and we may be able to offer useful advice.
Acknowledgements
This work was funded and supported by Calico Life Sciences LLC and we would like to acknowledge the fantastic research environment that has been created here by the senior staff. We would also like to specifically thank the yeast and worm groups within the company for helping prepare and provide the biological samples presented.
Special praise is given to the business development team at Calico for pursuing and arranging all of our external collaborations. In this regard we would like to specifically acknowledge our close collaborations with Nikon and Leica who made it possible to create a high quality instrument design and we look forward to future projects with these partners.
We also thank Physik Instumente, Lumencor and PCO for their detailed assistance in using their products, and our local suppliers Zera Development and Mark Optics for their high quality work and readily available technical support.
Appendix
Additional details and discussion can be found in the appendix, which is also referenced via hyperlinks throughout this article.